Malattia di Moyamoya: sintomi e terapia

Scritto e verificato il dottore Leonardo Biolatto
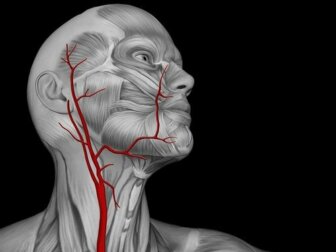
La malattia di Moyamoya è una patologia che colpisce le arterie. Per l’esattezza, si tratta di un disturbo legato all’angiogenesi, ovvero alla formazione dei vasi sanguigni. Colpisce le arterie carotidi interne nel tratto d’ingresso al cranio.
Anatomicamente, le arterie carotidi nascono come due vasi vicini al cuore. Si dirigono verso il collo, dove si dividono in due: arterie carotidi interne e arterie carotidi esterne. Dal collo, ogni coppia di vasi entra nel cranio trasportando il sangue pompato dal cuore al cervello. Si può facilmente intuire, dunque, che una patologia che interessa questi vasi è potenzialmente letale.
Nella malattia di moyamoya le arterie carotidi interne si restringono, riducendo l’afflusso di sangue al cervello. Giacché il corpo umano ha tra le sue priorità quella di garantire una buona irrorazione cerebrale, sviluppa una rete di piccoli vasi sanguigni per ovviare all’ostruzione.
Questi nuovi vasi, svolgono la funzione di creare una via accessoria alla risalita del sangue dal cuore. I piccoli vasi collaterali situati alla base del cranio, se osservati con metodi diagnostici complementari, emulano il fumo di sigaretta nell’aria.
“Moya-moya” in giapponese significa “nuvola di fumo”. Da qui l’etimologia del nome della malattia. È anche il termine che descrive il fumo che esce dai vulcani. Attraverso le immagini angiografiche, gli scienziati Susuki e Takaku hanno dato il nome a questa malattia nel 1969.
Distribuzione della malattia di Moyamoya
La malattia di Moyamoya presenta due picchi d’incidenza: nei bambini e negli adulti di mezza età. La prima fascia d’età più colpita riguarda i bambini tra i cinque e i dieci anni. Il secondo gruppo è composto da persone tra i trenta e i cinquanta anni.

Sebbene si siano verificati casi in tutto il mondo, pur con bassa frequenza, si può affermare che l’area più colpita sia il sud-est asiatico. Paesi come Giappone, Cina e Corea concentrano il maggior numero di casi globali. In Giappone, ad esempio, l’incidenza è di uno ogni trentaduemila abitanti.
Sempre nel sud-est asiatico, sono stati condotti una serie di studi che hanno rilevato che su un campione di duemila giapponesi adulti che non presentano sintomi della patologia, almeno uno presenta immagini cerebrali compatibili con la malattia di moyamoya.
Può interessarvi anche: inquinamento e cervelli dei bambini
Fattori di rischio
L’eziologia della malattia non è a tutt’oggi ben chiara. Si sospetta che l’origine sia genetica e che, pertanto, l’ostruzione arteriosa abbia una componente ereditaria. A ogni modo, si conoscono cinque fattori di rischio che ne aumentano la predisposizione:
- Storia familiare: quando un membro della famiglia ha avuto la malattia di moyamoya, le probabilità che un altro familiare diretto ne soffra aumentano di 40 volte.
- Discendenza asiatica: probabilmente l’associazione avviene per motivi genetici. I soggetti nati o provenienti dal sud-est asiatico hanno più probabilità di contrarre il disturbo rispetto al resto delle popolazioni mondiali.
- Sesso femminile: la malattia è più frequente tra le donne.
- Minori di quindici anni: l’età pediatrica, come spiegato sopra, è una delle fasce più colpite.
- Altre malattie: alcuni casi di moyamoya sono stati riscontrati in combinazione ad altre patologie, come la sindrome di Down.
Sintomi della malattia di Moyamoya
La prima manifestazione clinica è l’ictus. Questo perché l’ostruzione arteriosa non lascia fluire sufficiente sangue al cervello o perché i vasi accessori sono deboli e si rompono in presenza di uno sforzo. Sia l’ischemia che l’emorragia possono avere esito letale.
L’ictus può dare segnali di avvertimento. Nei bambini, di solito si manifestano delle convulsioni ripetute che rispondono alla carenza di una sufficiente irrorazione cerebrale. Negli adulti può verificarsi un attacco ischemico transitorio che si esprime con emiparesi o emiplegia.

I seguenti sintomi sono dovuti alla scarsa irrorazione del cervello:
- Mal di testa
- Disturbi visivi
- Problemi di linguaggio
- Movimenti involontari
In età pediatrica, l’evento scatenante più frequente è il pianto. Quando i bambini piangono intensamente, iperventilano. L’iperventilazione presuppone una variazione nell’afflusso di sangue al cervello, che è regolato dalla costrizione e dalla dilatazione dei vasi sanguigni, andando incontro a una possibile rottura di questi ultimi.
Leggete anche: Le 4 malattie cerebrovascolari più comuni
Diagnosi e terapia
L’unico modo per diagnosticare la malattia di Moyamoya è attraverso le immagini diagnostico-strumentali. L’angiografia e l’angio-risonanza magnetica sono gli studi più rigorosi nel rilevare tale patologia. Attraverso le immagini ottenute, sarà possibile valutare la riduzione del lume arterioso nelle carotidi interne. Il trattamento si articola in due modi:
- Un’opzione è quella di tipo farmacologico. I farmaci non ne arrestano il decorso, ma contribuiscono a ridurre il rischio di ictus. Vengono somministrati anticoagulanti, vasodilatatori e antiaggreganti piastrinici.
- La seconda opzione è il trattamento chirurgico. L’intervento è indicato nei pazienti che hanno già subito un accidente cerebrovascolare e che, in presenza o meno di strascichi, sono più esposti a una recidiva.
Tutte le fonti citate sono state esaminate a fondo dal nostro team per garantirne la qualità, l'affidabilità, l'attualità e la validità. La bibliografia di questo articolo è stata considerata affidabile e di precisione accademica o scientifica.
- Scott RM: Moyamoya Syndrome, a surgically treatable cause of stroke in pediatric patient. Clin Neurosurg 2000; 47: 378-84.
- González-Rabelino G, Campistol J, Navarro-Balbuena R, Capdevila-Cirera A, Solá-Martinez T. Moyamoya en la población infantile. Análisis de una serie occidental y revisión de la literatura. Rev Neurol. 2008; 46 (7): 385-91.
- Satoshi Kuroda MD, Nobuo Hashimoto MD PhD, Takashi Yoshimoto MD PhD: Radiological Findings, Clinical Course, and Outcome in Asymptomatic Moyamoya Disease. Stroke 2007; 38: 1430-5.
- Kuroda S, AMORE Study group. Asymptomatic moyamoya disease: literature review and ongoing AMORE study. Neurol Med Chir (Tokyo). 2015; 55 (3): 194-8.
Questo testo è fornito solo a scopo informativo e non sostituisce la consultazione con un professionista. In caso di dubbi, consulta il tuo specialista.