
La sindrome di Turner è un’alterazione genetica che colpisce i cromosomi sessuali. È caratterizzata dall’assenza completa o parziale di uno dei due cromosomi X, quindi interessa solo il sesso femminile. Il suo cariotipo è, pertanto, 45X.
È l’unica malattia in cui un embrione monosomico può restare vitale. La monosomia (mancanza di uno dei cromosomi di una coppia di omologhi) degli autosomi, ossia dei cromosomi non sessuali è, invece, incompatibile con la vita. Anche così, il 99% degli embrioni con cariotipo 45X viene abortito in modo naturale nel primo trimestre.
L’assenza del cromosoma Y determina il sesso genetico femminile. La presenza di un singolo cromosoma X, invece, porta a un mancato sviluppo delle caratteristiche sessuali femminili primarie e secondarie.
Le statistiche
Statisticamente, circa il 50% delle pazienti con sindrome di Turner ha il cariotipo 45X, con perdita totale del secondo cromosoma. Il 15% delle pazienti presenta solo una perdita parziale del secondo cromosoma. In questo caso, l’alterazione può interessare il braccio lungo oppure il braccio corto del cromosoma.
Nel resto dei casi si parla di mosaicismo. I mosaicismi sono caratterizzati dalla presenza, nello stesso individuo, di due o più linee cellulari o patrimoni genetici diversi. In questo gruppo di persone, dunque, convivono cellule geneticamente normali (46XX) e cellule geneticamente modificate (45X).
Vi consigliamo di leggere: La Sindrome di Down potrebbe avere i giorni contati
Cromosomi sessuali in condizioni normali
In condizioni normali, l’essere umano possiede 23 coppie di cromosomi. 22 di queste sono chiamate autosomi o cromosomi non sessuali, la coppia restante è costituita dai due cromosomi sessuali. I cromosomi sessuali, ereditati rispettivamente dal padre e dalla madre, determinano il sesso del nascituro. In questo modo:
- La presenza di due cromosomi X (46XX) determina il sesso genetico femminile. Porta allo sviluppo di caratteristiche sessuali femminili primarie e secondarie.
- La presenza di un cromosoma Y (46XY) determina il sesso genetico maschile. Comporta lo sviluppo di caratteristiche sessuali maschili, primarie e secondarie.
La sindrome di Turner: cosa succede?
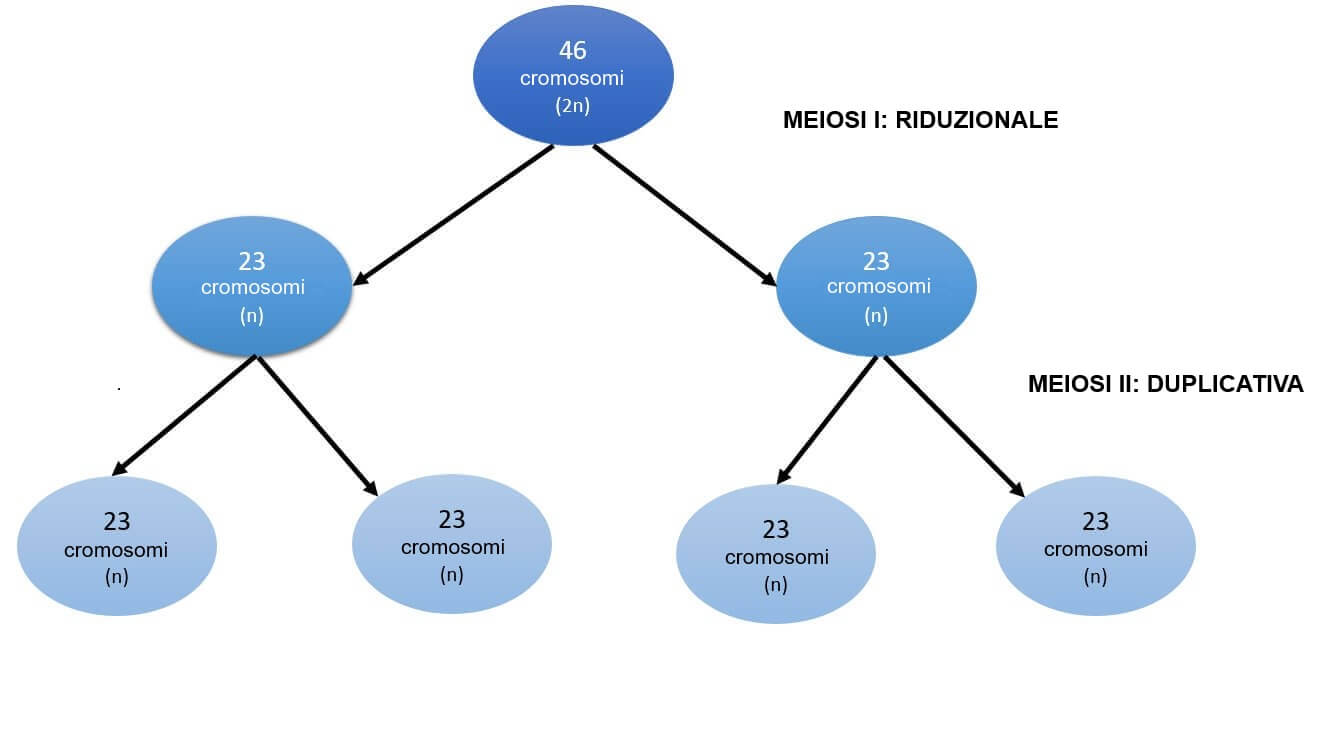
Prima di spiegare l’origine di questa alterazione cromosomica, è importante rivedere alcuni aspetti della divisione cellulare delle cellule riproduttive (gameti).
Il processo si chiama meiosi e si svolge in due fasi chiamate meiosi I e meiosi II. L’obiettivo finale è quello di produrre cellule figlie con metà del patrimonio genetico delle cellule originali.
Si parte da una cellula madre con 23 coppie di cromosomi. Dopo la meiosi I, si hanno due cellule figlie, composte da 23 cromosomi ciascuna. Ognuna di queste cellule, durante la meiosi II, origina a sua volta altre due cellule con corredo genetico di 23 cromosomi. Da una cellula madre diploide, dunque, si ottengono quattro cellule figlie aploidi.
Perché la cellula può perdere un cromosoma?
Secondo la teoria più accreditata, la perdita del secondo cromosoma, X o Y, si verifica dopo il concepimento. Si tratterebbe, quindi, di una falla nella divisione cellulare.
L’assenza del cromosoma Y comporta lo sviluppo di caratteristiche sessuali femminili. Pertanto, anche se originariamente la cellula era di tipo XY, la perdita del cromosoma determinerà la nascita di una bambina.
Sintomi della sindrome di Turner
Aspetto fisico
- Ritardo nella crescita durante l’infanzia e bassa statura in età adulta.
- “Volto a sfinge“: ponte nasale appiattito, orecchie sporgenti con lievi malformazioni e palpebre cadenti.
- Collo più corto e più largo del normale e capelli con attaccatura molto bassa.
- Presenza frequente di nei pigmentati.
- Torace ampio, con seno poco sviluppato, dall’aspetto infantile e più separato del solito.
- Caratteri sessuali secondari poco sviluppati o infantilismo sessuale. Assenza di sviluppo del seno e dei peli pubici e mancato arrotondamento dei fianchi. Tutto ciò a causa dell’assenza degli estrogeni.
- Sono frequenti le malformazioni ossee, come il cubito valgo (deviazione dell’avambraccio verso l’esterno).
Leggete anche: Origine genetica del sesso: i cromosomi X e Y
Malformazioni interne
- Sono molto comuni le cardiopatie congenite, come la coartazione aortica (restringimento dell’aorta) o le malformazioni valvolari.
- Anche le malformazioni renali sono piuttosto frequenti; la più comune è il rene a ferro di cavallo.
- Disgenesie gonadiche. Le ovaie non sono ben sviluppate con presenza comune di gonadi a striscia. La sindrome di Turner comporta un quadro di ipogonadismo ipergonadotropo. Ciò significa che, sebbene ci siano tutti gli stimoli necessari per la produzione degli estrogeni, questi non vengono prodotti a causa di un’alterazione delle ovaie.
- Amenorrea (assenza di mestruazioni) e infertilità. Non si verifica sempre e, in effetti, una piccola percentuale delle pazienti è fertile. Questa è una delle caratteristiche più importanti della sindrome di Turner.
Diagnosi

La sindrome di Turner ha una diagnosi difficile da determinare. La maggior parte delle bambine affette da questa malattia non ha caratteristiche molto marcate; ovvero sono fenotipi lievi, poiché i casi più marcati sono abortiti in modo naturale.
Circa un terzo dei casi viene diagnosticato in fase neonatale. Il sospetto si basa sulla presenza di tratti fisici caratteristici e sintomi di malattie cardiache. Un altro terzo di solito viene diagnosticato durante l’infanzia, principalmente a causa della bassa statura
L’ultimo terzo viene diagnosticato durante l’adolescenza. In questi casi, alla bassa statura si aggiunge l’infantilismo sessuale e l’impossibilità di raggiungere la fase della pubertà.
Trattamento
La terapia ormonale sostitutiva a base di estrogeni aiuta a sviluppare le caratteristiche sessuali, ma non corregge l’infertilità. Durante l’infanzia si consiglia di somministrare alle bambine l’ormone della crescita che aiuta, più o meno, a normalizzare la statura.
È possibile avere figli con la sindrome di Turner?
Sì, è possibile, ma solo una piccola percentuale di donne affette da sindrome di Turner è fertile.
In caso di sterilità, situazione più comune, è necessario ricorrere a tecniche di fecondazione in vitro con ovuli provenienti da donatrici. L’ovulo donato e fecondato in vitro viene successivamente impiantato nell’utero materno. Una gravidanza del genere richiede, naturalmente, un controllo molto più rigoroso di una normale gestazione.
Bibliografia
Tutte le fonti citate sono state attentamente esaminate dal nostro team per garantirne la qualità, affidabilità, rilevanza e validità. La bibliografia di questo articolo è stata considerata affidabile e di precisione accademica o scientifica.
- Wyss D, DeLozier CD, Daniell J, Engel E. Structural anomalies of the X chromosome: personal observation and review of non-mosaic cases. Clin Genet. 2009; 21(2):145-59.
- Jacobs PA, Betts PR, Cockwell AE, Crolla JA, Mackenzie MJ, Robinson DO, et al. A cytogenetic and molecular reappraisal of a series of patients with Turner’s syndrome. Ann Hum Genet. 1999; 54(Pt 3):209-23.
- Lippe BM. Turner’s syndrome. In: Sperling M. Pediatric endocrinology. 3 ed. Philadelphia: Saunders; 2008.p. 387.
- Chagoyén Méndez, Esther María, Álvarez Montero, José Agustín, & Zúñiga Vaca, Carmen Isabel. (2017). Síndrome de Turner en una adolescente. MEDISAN, 21(6), 720-724. scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192017000600012&lng=es&tlng=es.