Pubblicato il 5 Settembre 2022 • Aggiornato il 13 Dicembre 2022
7 minuti di lettura
Il sarcoma di Ewing consiste nella formazione di tumori ossei, che manifestano con noduli, infiammazioni, debolezza ossea, rigidità, perdita di peso, affaticamento e persino fratture spontanee.
Le cause esatte non sono note, sebbene siano stati identificati fattori di rischio associati, come l’età o l’etnia. I trattamenti includono radioterapia e chemioterapia, tra gli altri.
Il tasso di sopravvivenza è compreso tra il 60 e il 70% dei pazienti.
Cos’è il sarcoma di Ewing?
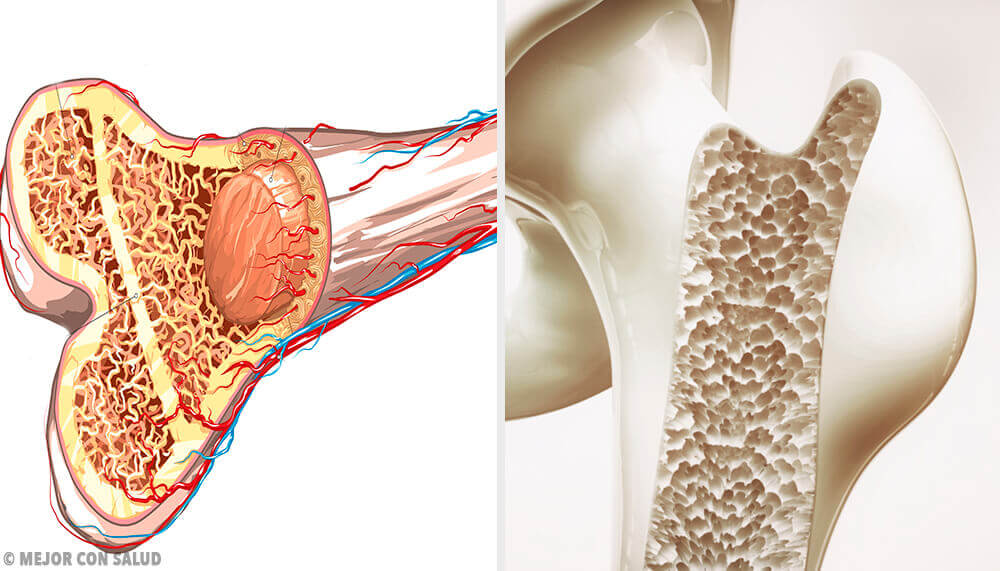
Il sarcoma di Ewing è un tipo di cancro piuttosto raro che attacca l’osso circostante o i tessuti molli, formando tumori. Questi possono diffondersi ad altre aree, creando delle metastasi.
Non è comune negli adulti. In generale, le persone colpite sono bambini e adolescenti, per lo più maschi. Soprattutto durante la pubertà, quando le ossa sono in piena crescita. La sua incidenza è stimata in 2,73 casi per milione di persone.
Sebbene possano avere manifestazioni simili, ci sono differenze tra l’osteosarcoma e il sarcoma di Ewing. In relazione alla frequenza, il primo è più comune. Nella loro origine, hanno origine da cellule diverse.
Il sarcoma di Ewing è più comune nelle ossa degli arti inferiori, in particolare nel femore. Così come nel bacino. Tuttavia, potrebbe apparire su altri siti.
Segni e sintomi includono quanto segue:
Dolore osseo: può essere intermittente e meno intenso di notte.
Gonfiore: sensazione di grumo, che può risultare morbido al tatto.
Molti di questi segni possono essere dovuti ad altre patologie. Tuttavia, è importante consultare un medico se li si verifica. Ci sono anche casi di persone con sarcoma di Ewing che non hanno sintomi.
Le ossa rappresentano il tessuto più colpito da questo sarcoma, soprattutto durante la pubertà.
Cause e fattori di rischio
Non è stato possibile determinare esattamente quali siano le cause del sarcoma di Ewing. È noto che questa patologia inizia quando le cellule subiscono cambiamenti nel loro DNA, facendole moltiplicare senza controllo.
Questi cambiamenti il più delle volte interessano un gene noto come EWSR1. Ciò si traduce in una massa tumorale di cellule che possono diffondersi, generando delle metastasi nelle altre ossa.
Inoltre, è noto che questa malattia non è ereditaria. Per quanto riguarda i fattori di rischio per il sarcoma di Ewing, ci sono i seguenti:
Età: sebbene possa verificarsi a qualsiasi età, la maggior parte dei casi riguarda persone di età compresa tra 10 e 20 anni.
Sesso: è più comune nei maschi.
Etnia: non è così comune tra le persone di etnia nera o asiatica, rispetto a quelle di origine europea.
La diagnosi del sarcoma di Ewing inizia con un esame fisico. Inoltre, il paziente oi suoi genitori vengono interrogati in merito ai sintomi, alla loro frequenza e durata. Da questo, possono essere raccomandati diversi test.
Prove di imaging
I test di imaging iniziano con i raggi X, che consentono di rilevare le masse tumorali evidenti. Viene eseguita anche la tomografia computerizzata, specialmente nell’area del torace, per vedere se c’è una diffusione ai polmoni.
Allo stesso modo, la risonanza magnetica può essere applicata per tali scopi. Infine, vengono eseguite la tomografia a emissione di positroni e la scintigrafia ossea.
Biopsia
Viene eseguita una biopsia dell’ago. In esso, un ago viene inserito attraverso la pelle, fino al tumore, per estrarre un campione. I campioni vengono inviati al laboratorio.
In alternativa, una biopsia chirurgica può essere eseguita dopo l’intervento chirurgico per rimuovere il tumore. Sebbene ci sia la possibilità di rimuovere solo una parte del tessuto attraverso una piccola incisione.
Altre analisi
Oltre a quelli citati, vengono eseguiti vari test di laboratorio:
Emocromocompleto.
Test del DNA: per determinare se ci sono mutazioni genetiche.
In base all’andamento della malattia, alle condizioni e all’età del paziente, l’équipe medica deciderà il protocollo da seguire. I trattamenti più comuni utilizzati per il sarcoma di Ewing sono descritti di seguito.
Chemioterapia
L’approccio è di solito iniziato con la chemioterapia. Potenti farmaci vengono combinati per distruggere le cellule tumorali.
I farmaci possono essere somministrati per via orale o endovenosa. Tra i più comuni ci sono ciclofosfammidi, doxorubicina, vincristina e dactinomicina.
Quando il tumore non si è diffuso, la chemioterapia viene effettuata ogni 2 settimane. Anche se è necessario un intervento chirurgico, il trattamento con questi farmaci può continuare dopo l’intervento chirurgico.
La chemioterapia ha spesso effetti collaterali, come perdita di capelli, perdita di appetito, nausea, vomito, affaticamento e aumento del rischio di infezione dovuto allo sviluppo di neutropenia.
La chemioterapia è aggressiva e ha molti effetti negativi. I bambini possono subire conseguenze a medio e lungo termine.
Chirurgia
In caso di intervento chirurgico, l’obiettivo è eliminare completamente le cellule tumorali. I casi più complicati arrivano all’amputazione di un arto.
Altre volte, il tumore viene rimosso senza amputare. Tuttavia, quando una parte del tessuto osseo viene persa, la funzione dell’arto ne risente.
Potrebbero essere necessari innesti, di tessuto osseo proprio o di donatore, nonché impianti (protesi in metallo o plastica).
Radioterapia
La radioterapia è anche un’opzione dopo l’intervento chirurgico, per distruggere le cellule tumorali rimanenti. In generale, aiuta a ridurre la crescita del cancro e ad alleviare i sintomi, come il dolore.
I suoi effetti collaterali includono affaticamento, reazioni cutanee e mal di stomaco. A lungo termine, può interferire con la crescita ossea e persino aumentare il rischio di cancro secondario.
Altre procedure
Oltre a quelle citate, esistono altre procedure per il trattamento del sarcoma di Ewing. Tra questi vi è il trapianto di midollo osseo, allogenico o autologo.
Può essere fatto alternando chemioterapia o radioterapia. La sua efficacia è ancora oggetto di studio.
Monitoring e assistenza
Anche quando il trattamento è stato completato, i pazienti con sarcoma di Ewing devono continuare a essere valutati e monitorati. Al riguardo, si formulano le seguenti raccomandazioni:
Le radiografie dovrebbero essere eseguite ogni 6 mesi per 5 anni. Poi ogni anno per il resto della loro vita.
Inoltre, scansioni polmonari e ossee regolarmente.
D’altra parte, devonoandare in terapia fisica dopo l’intervento chirurgico, per aiutare a ritrovare la mobilità o imparare a usare un arto se ci sono impianti.
Ci sono anche servizi di supporto psicologico per aiutare in caso di perdita dell’arto.
Prognosi e complicazioni
A volte, il corpo non risponde al trattamento e il sarcoma non può essere controllato, passando allo stadio di malattia avanzata o terminale. In ogni caso, le procedure hanno generalmente successo in un numero elevato di casi.
Le cifre su questo variano. Si stima un tasso di sopravvivenza a 5 anni del 75% per i minori di 15 anni.Questa percentuale diminuisce leggermente con l’età; fino al 58% per gli adolescenti. Lo stesso accade quando ci sono metastasi.
Potrebbe esserci una remissione permanente o solo temporanea, causando la ricomparsa del cancro. C’è più possibilità di recidiva durante i primi 2 anni dopo aver terminato il trattamento.
Le recidive tardive (fino a 5 anni dopo) sono più comuni con il sarcoma di Ewing che con altri tipi di cancro. Quando ritorna, può essere più difficile da trattare, poiché i farmaci che erano usati per la chemioterapia non possono essere nuovamente indicati a causa del rischio di tossicità.
Bibliografia
Tutte le fonti citate sono state attentamente esaminate dal nostro team per garantirne la qualità, affidabilità, rilevanza e validità. La bibliografia di questo articolo è stata considerata affidabile e di precisione accademica o scientifica.
Hernández González E, Mosquera Betancourt G, Quintero Martínez O, Hernández Cabezas I. Sarcoma de Ewing. AMC. 2013; 17(5): 623-640.
Jiménez Soto D. Sarcoma de Ewing en Pelvis. Revisión bibliográfica. San José: Universidad de Costa Rica, 2019.
Khanna N, Pandey A, Bajpai J. Metastatic ewing’s sarcoma: Revisiting the “Evidence on the Fence”. Indian J Med Paediatr Oncol. 2017; 38: 173-181.
Sánchez J. Lactato deshidrogenasa. Bol. Hosp. San Juan de Dios. 1999; 46(3): 182-187.
Muñoz Villa A. Tumores óseos. Rabdomiosarcomas. Pediatr Integral 2016; XX (7): 458 – 464.
Thowinson-Hernández M, Hernández-Martínez A. Neutropenia febril inducida por quimioterapia e infecciones asociadas: una revisión de la literatura. Gaceta Mexicana de Oncologia. 2019; 18: 1-6.
Horowitz M, Kinsella T, Wexler L, et al. Total-body irradiation and autologous bone marrow transplant in the treatment of high-risk Ewing’s sarcoma and rhabdomyosarcoma.. Journal of Clinical Oncology. 1993; 11(10):1911-1918.